
Studiendesign
Auf dieser Seite finden Sie alle Informationen zum Studiendesign und den erhobenen Datensätzen in NAPKON.
Studieninterventionen
NAPKON ist eine Kohortenstudie mit drei Kohorten: der Sektorenübergreifenden Plattform (SÜP), der Hochauflösenden Plattform (HAP) und der Populationsbasierten Plattform (POP). Die SÜP rekrutiert stationäre und ambulante Patient:innen an Universitätskliniken, Nicht-Universitären Kliniken und Arztpraxen; die HAP rektuiert ausschließlich stationäre Patient:innen an ausgewählten Universiätskliniken; die POP rekrutiert ausschließlich ambulante Patient:innen über Einladungen durch die Gesundheitsämter an Universitätskliniken. Weiterführende Informationen zu den Kohorteneigenschaften können Sie auf der Projektseite und auf unserer Standortseite nachlesen. Eine ausführliche Beschreibung zum Studiendesign von NAPKON bietet die Übersichtspublikation.
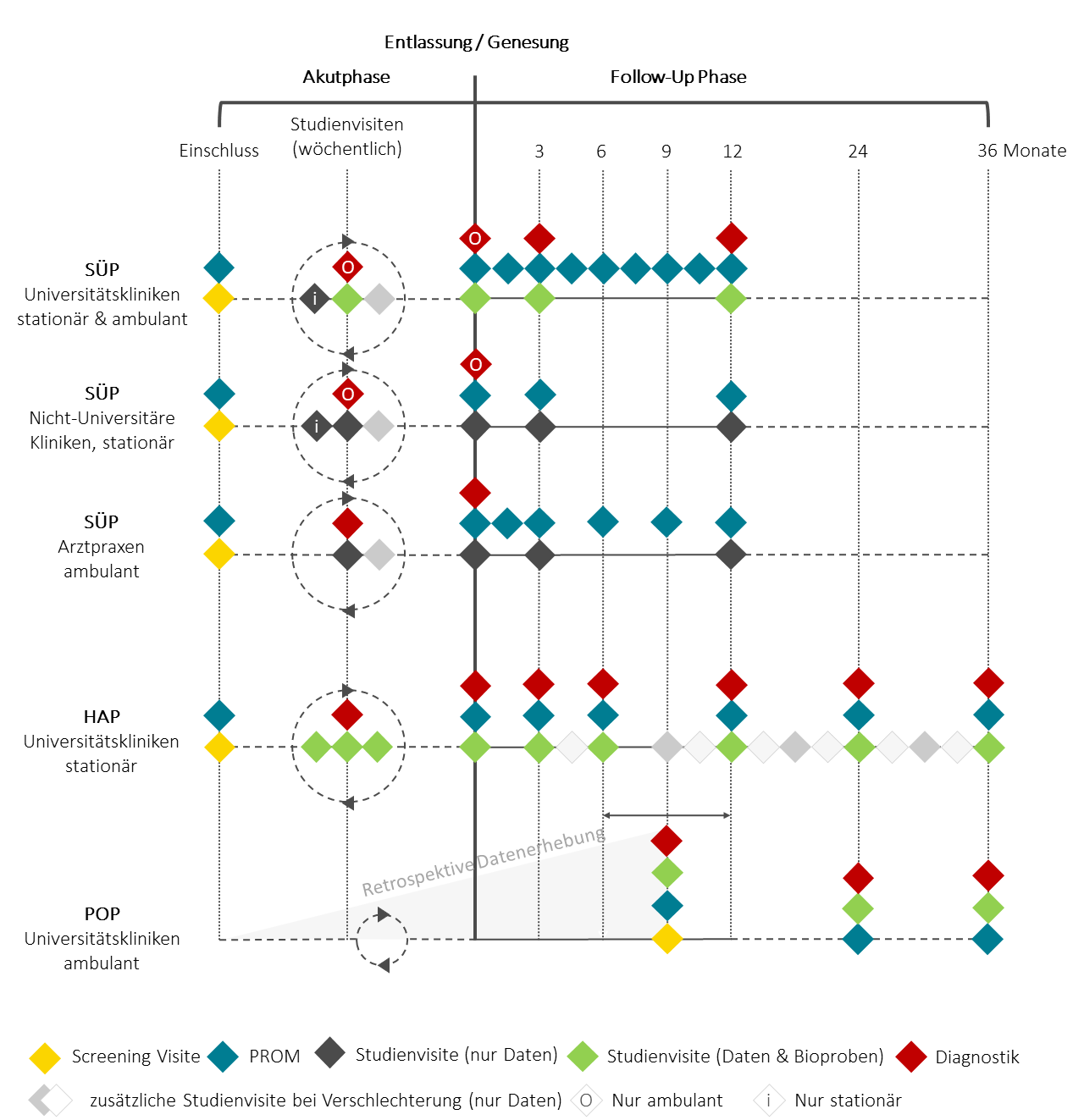
Das Visitenschema gibt ihnen einen Überblick über die Art und Frequenz der durchgeführten Studieninterventionen.
Letzte Aktualisierung: Februar 2023
Datensätze in NAPKON
In diesem Abschnitt erhalten Sie eine Übersicht über die erhobenen Datensätze in NAPKON.
Allgemeines
In NAPKON werden klinische Daten, Bilddaten und Bioproben erhoben. Die individuellen Entnahmezeitpunkte können Sie dem Visitenschema (siehe oben) entnehmen. Der klinische Teil des NAPKON Datensatzes setzt sich zusammen aus den kohorten-spezifischen Kerndatensätzen und kohorten-spezifischen zusätzlichen Datenelementen. Die Kohortendatensätze werden um ebenfall kohorten-spezifische Bilddaten und Bioproben erweitert.
Im Rahmen eines Unterarbeitspakets in NAPKON wird der GECCOplus Datensatz erstellt. Dieser ist ein harmonisierter Datensatz über die klinischen Daten aller drei Kohorten.
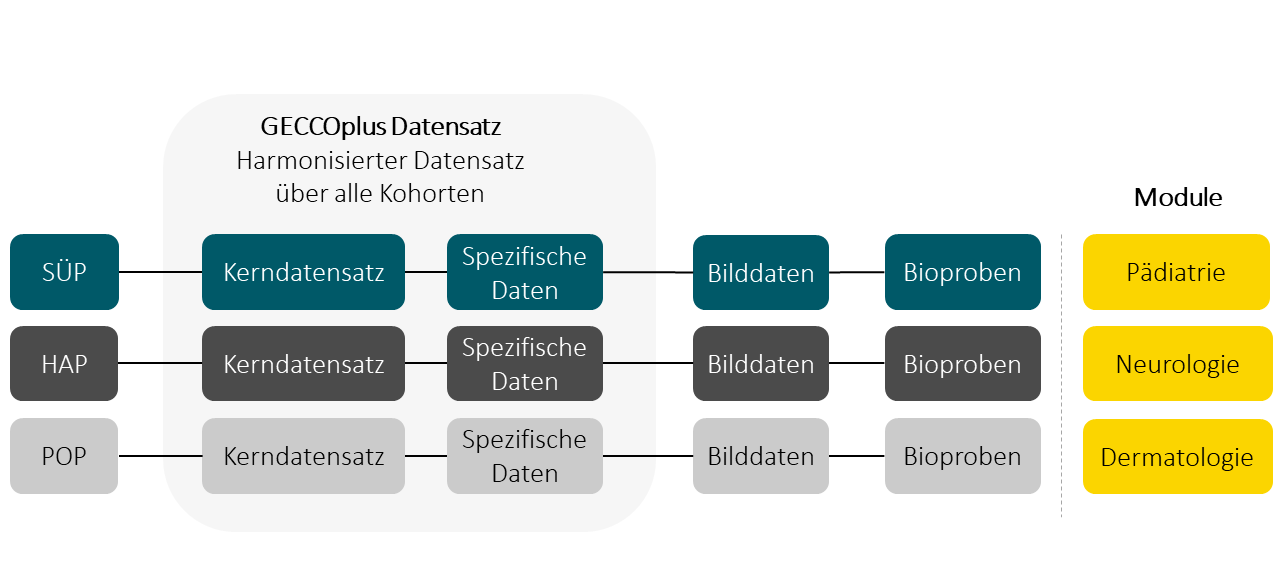
Der GECCOplus Datensatz
In NAPKON bildet der GECCO Kerndatensatz den Minimaldatensatz und ist in den Kohortendatensätzen von SÜP, HAP und POP mit Einschränkungen enthalten. Eine ausführliche Beschreibung des GECCO Kerndatensatzes finden sie hier.
Er wurde durch ein klinisches Expertengremium gemeinsam in einem beschleunigten Verfahren definiert. Der konsentierte Datensatz enthält gemeinsam definierte Datenelemente zur Erfassung von wichtigen patient:innenbezogenen klinischen Daten im Rahmen der COVID-19 Pandemie.
Eine Erweiterung des Kerndatensatzes und Harmonisierung der vollständigen Kohortendatensätze ist der GECCOplus-Datensatz. Er ist die Schnittmenge der Datenelemente aller drei NAPKON-Kohortenplattformen SÜP, HAP und POP.
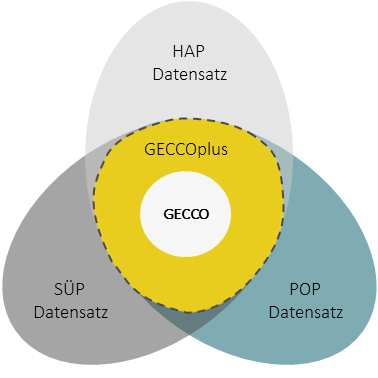
Da die Kohortendatensätze über die Zeit stetig aktualisiert werden – Datenparameter entfernt, hinzugefügt oder angepasst werden – wird auch der GECCOplus entsprechend überprüft und adjustiert. Für den Prozess der Aktualisierung wurde die AG GECCOplus Qualitätssicherung gegründet. Sie vereint u.a. die Expertise der Kohortenplattformen, der Transferstelle und der ECU und trifft sich auf regelmäßiger Basis um den Prozess voran zu treiben.
Der NAPKON PUF Datensatz
Der NAPKON PUF (Public Use File) Datensatz ist ein öffentlich zugänglicher Datensatz welcher Patient:innendaten aller drei Kohorten enthält. Die Daten sind qualitätsgesichert und anonymisiert. Weitere Informationen zum NAPKON PUF Datensatz finden Sie hier.
Bilddaten in NAPKON
In diesem Abschnitt finden Sie alle wichtigen Informationen zu den Bilddaten in NAPKON.
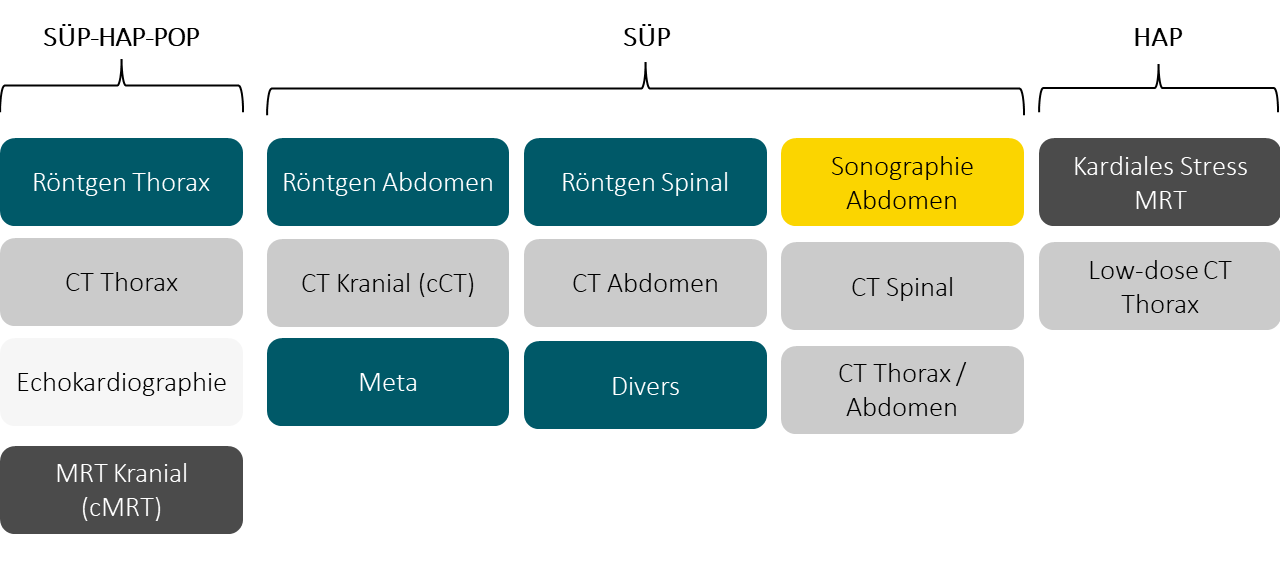
In NAPKON werden Bilddaten verschiedenen Typs erhoben. Dabei werden einige Typen in allen drei Kohorten, andere in nur ausgewählten Kohorten erhoben. Die erhobenen Bilddaten werden über das NUKLEUS DICOM Managementsystem (NUKLEUS DIMA) verwaltet und für den Use & Access Prozess ausgewählt.
Bioproben in NAPKON
In diesem Abschnitt finden Sie alle wichtigen Informationen zu den Bioproben in NAPKON.
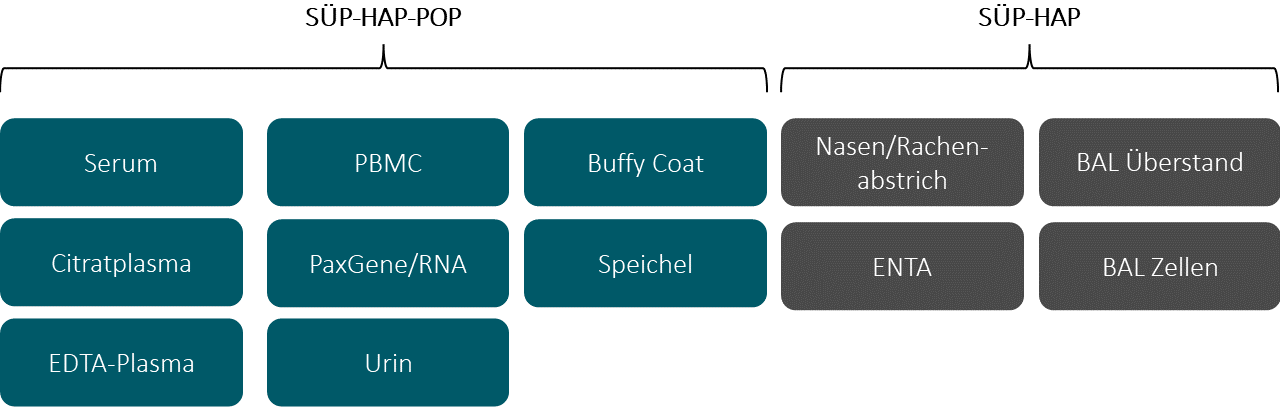
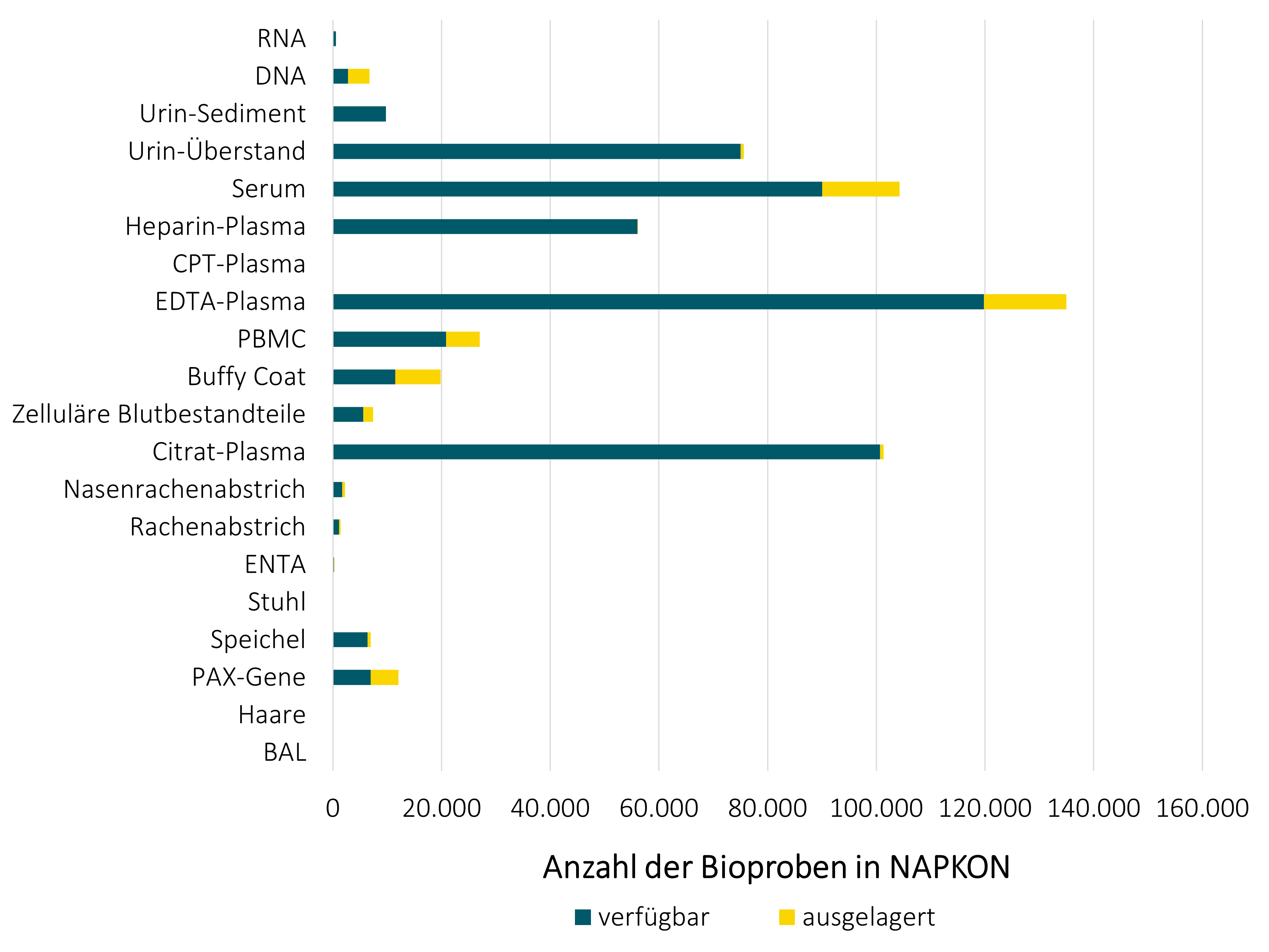
In NAPKON werden an allen beteiligten Standorten und Biobanken Bioproben verschiedenen Typs (siehe Grafik unten) nach einheitlichen Standards mit hoher Qualität lokal verarbeitet und gelagert. Sämtliche Standorte werden vom NUKLEUS Bioprobenkern (NUKLEUS BCU) regelmäßigen Audits unterzogen, um eine gleichbleibende Qualität zu gewährleisten. Die Bioproben werden zum Großteil in allen drei Kohorten (HAP, SÜP, POP), erhoben Nach Genehmigung von Forschungsprojekten im Use und Access Verfahren werden die Bioproben vom BCU von den lokalen Biobanken angefordert und nach Trockeneistransporten zwischengelagert und anschließend Qualitäts-kontrolliert. Nach erfolgter Qualitätskontrolle werden die Bioproben den jeweiligen Analyselaboren zugeschickt.
Der NUKLEUS Bioprobenkern (BCU) arbeitet intensiv mit weiteren NUKLEUS Infrastrukturen wie dem Biosample Information Management System (BIMS), der Epidemiology Core Unit (ECU), dem Interaktionskern (ICU), dem Klinischen Datenmanagement (CDM), der Transferstelle (TO), sowie der Treuhandstelle (THS) zusammen. Der BCU hat einen zentralen Überblick über Gewinnung und Nutzung der NAPKON Bioproben. Insgesamt wurden bis Ende Juli 2023 566.565 Primärproben und Aliquote generiert und 57043 Bioproben an 26 Forschungsprojekte ausgegeben.
Stand der Bioproben: 31.07.2023
Das Projekt SAPCRiN
In NAPKON wurde ein auf der Analyse von Bioproben basierendes Projekt verankert: SAPCRiN – Sample Analysis for Post COVID Research in NAPKON. Es dient dazu die molekularen Veränderungen zu charakterisieren, die während der COVID-19-Infektion und Genesung auftreten und ob diese Veränderungen mit der Entwicklung des Post-COVID-Syndroms (PCS) zusammenhängen.
Nähere Informationen zum Projekt SAPCRiN finden Sie hier.
Molekulare Analysen
Folgende molekulare Analysedaten werden im Rahmen von NAPKON v1.0 für insgesamt 2520 (1217 SUEP, 300 HAP, 1003 POP) Patient:innen generiert und können über den normalen UAC-Prozess beantragt werden.
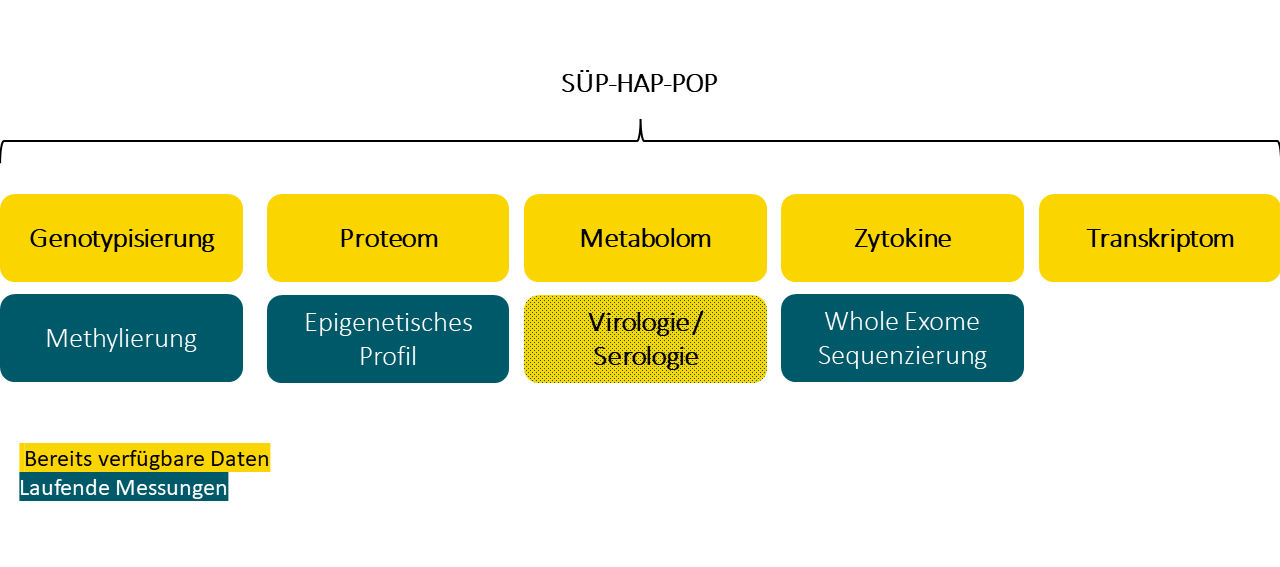
+++++ Verfügbare Daten sind schwarz hinterlegt. Noch nicht verfügbare Daten sind grau hinterlegt. +++++
Genotypisierung
Genotyp-Daten werden bei Dr. Christian Gieger in der Abteilung Molekulare Epidemiologie am Helmholtz Zentrum München erhoben.
Verwendet wird dabei der Infinium Global Screening Array (GSA)-24 v3.0 BeadChip der Firma Illumina (https://emea.illumina.com/products/by-type/microarray-kits/infinium-global-screening.html), mit dem bereits über 200.000 Proben in Kiel (IKMB, CAU), Bonn (Life &Brain) und München (HMGU) durch das GSA Konsortium genotypisiert wurden. Der GSA Array ist ein klinisch relevanter, multi-ethnischer, genomweiter Array, der Hochdurchsatzgenotypisierung ermöglicht. Der GSA deckt 700,000 genetische Varianten ab, einschließlich 600,000 prädiktiver Marker für maximale genomische Abdeckung und Imputationsgenauigkeit und 50,000 klinisch relevante Marker, mit denen direkt auf pathogene und vermutlich pathogene genetische Variationen getestet werden kann. Die klinischen Marker basieren auf den Datenbanken ClinVar, NHGRI, PharmGKB, und ExAC.
Proteom
Proteom-Daten werden in der Arbeitsgruppe von Prof. Markus Ralser am Institut für Biochemie, Charité Universitätsmedizin Berlin erhoben.
Die Serum/Plasma-Proteomik gilt als Technologie mit großem Potential für die Präzisionsmedizin (Prasad et al., J Pharm Sci 2017: PMID 27939376; Van Eyk et al., J Proteome Res 2019: PMID 30296097).
Die verwendete Technologie kombiniert Hochdurchflusschromatographie mit Scanning SWATH, einer für den Hochdurchsatz entwickelten massenspektrometrischen Erhebungsmethode (Messner et al., Cell Syst 2020: PMID 32619549; Demichev et al., Nat Methods 2020: PMID 31768060; Messner et al., Nat Biotechnol 2021: PMID: 33767396)
Mithilfe dieser Plattform wurden bereits Proben der Pa-COVID-19 Kohorte analysiert und potenzielle Biomarker identifiziert, die in Abhängigkeit vom COVID-19 Schweregrad differenziell exprimiert werden (Messner et al., Cell Syst 2020: PMID 32619549; Messner et al., Nat Biotechnol 2021: PMID: 33767396).
Metabolom
Metabolom-Daten werden an den Standorten des NMR-Konsortiums (Greifswald, Lübeck, Oldenburg und Tübingen) erhoben.
Verwendet wird die NMR-Spektroskopie. Mit der 1H-NMR-Spektroskopie besteht die Möglichkeit, eine große Anzahl von Substanzen in biologischen Flüssigkeiten zu identifizieren und zu quantifizieren (K. Suhre et al., Nat Genet 2011; PMID: 21572414).
Die Zahl der identifizierten Substanzen in den Bibliotheken wächst aktuell ständig. Im Vergleich zu anderen Messverfahren ist die Quantifizierung definierter Metabolite auf relativ einfache Weise möglich, da ein Universalstandard ausreicht und nicht für alle Metabolite spezifische Standards benötigt werden. Außerdem erfasst die NMR-Spektroskopie aus vielen Substanzen und Spektren zusammengesetzte metabolische Signaturen, die zur Charakterisierung von Patientengruppen eingesetzt werden können (Raffler et al., PLoS Genet 2015; PMID: 26352407; M. Pietzner et al., J Clin Endocrinol Metab 2018; PMID: 30060179; A. Kaul et al., Nutrients 2018; PMID: 30463274;Friedrich et al.,Metabolomics 2015;11:1405-15; M. Pietzner et al., BMC Med 2017; PMID: 29187192; T. Lange et al., J Clin Endocrinol Metab 2018; PMID: 29546278).
In Zusammenhang mit SARS-CoV-2-Infektionen wurden unter Anwendung der NMR-Spektroskopie bereits zahlreiche Assoziationen beschrieben, die unter anderen den Fettstoffwechsel, Aminosäuren (Kimhofer et al., J Proteome Res. 2020; PMID: 32806897), Amine (Lawler et al., J. Proteome Res 2021; PMID: 33724837) und weitere Metabolite (Thomas et al., JCI Insight 2020 Jul; PMID: 32559180; Meoni et al., PLoS Pathog 2021; PMID: 33524041) betreffen.
Zytokine
Zytokin-Daten werden bei Prof. Dr. Matthias Nauck an der Universitätsmedizin Greifswald erhoben.
Hier kommt ein Multiplex-ELISA zum Einsatz, der bereits seit Jahren etabliert ist und mit dem zahlreiche Großprojekte, wie z.B. SHIP (Study of Health in Pomerania) untersucht wurden.
„Zytokine“ ist ein allgemeiner Begriff für eine heterogene Gruppe löslicher Proteine und Peptide, die sowohl in physiologischen als auch pathologischen Situationen immunmodulierend wirken. Die Gruppe der Zytokinproteine umfasst Interleukine, Interferone, koloniestimulierende Faktoren und Chemokine.
Eine detaillierte Auflistung der gemessenen Parameter steht für sie zum Download bereit. Bei Fragen zu der Parameter Tabelle wenden Sie sich bitte an das LIMS-Team: num-lims@med.uni-greifswald.de
Mit Blick auf die gegenwärtige Pandemie wurden inflammatorische Zytokin-Signaturen veröffentlicht, die eine Vorhersage des Schweregrades einer COVID-19 Erkrankung sowie die Überlebenswahrscheinlichkeit erlauben (Del Valle et al., Nat Med 2020; PMID: 32839624).
Transkriptom
Das Transkriptionsprofil in aufgereinigten Blutzellen (PBMCs) wird in der Arbeitsgruppe von Prof. Dr. Soni Pullamsetti an der Justus-Liebig-Universität in Gießen bestimmt. Die Bestimmung erfolgt durch RNA-Sequenzierung auf Next Generation Sequenziergeräten der Firma Illumina. Transkriptomanalysen ermöglichen quantitative Aussagen über Genexpressionsmuster und qualitative Aussagen über Nukleotidveränderungen im kodierenden und nicht-kodierenden Bereich.
Methylierung
Daten zur genomweiten Methylierung werden bei Dr. Christian Gieger in der Abteilung Molekulare Epidemiologie am Helmholtz Zentrum München erhoben.
Zum Einsatz kommt hier der Infinium Methylation EPIC Array v1.0 BeadChip der Firma Illumina (https://emea.illumina.com/products/by-type/microarray-kits/infinium-methylation-epic.html).
Diese Technologie basiert auf der Behandlung von genomischer DNA mit Natriumbisulfit, welches zu einer Umwandlung von Cytosin in Uracil führt. Modifiziertes 5-Methylcytosin wird hingegen nicht umgewandelt.
Durch die anschließende Genotypisierung der einzelnen CpG-Stellen unter Verwendung von Sonden auf einem Microarray, kann der Methylierungsgrad bestimmt werden. Mit dem EPIC Array kann der Methylierungsstatus von 850.000 CpG Loci erfasst werden. Davon verteilen sich 500.000 CpGs auf 21.231 Gene (~ 99% der UCSC RefSeq-Gene), sowie weitere über 350.000 CpGs in Regionen, welche von FANTOM5 und dem ENCODE-Projekt als potenzielle Enhancer identifiziert wurden.
Epigenetisches Profil
Zur weiteren Bestimmung des epigenetischen Profils werden Hochdurchsatzverfahren (NGS) in der Arbeitsgruppe von Prof. Dr. Soni Pullamsetti an der Justus-Liebig-Universität in Gießen an Blutzellen (PBMCs) durchgeführt.
- Untersuchung der Chromatin Zugänglichkeit mittels Sequenzierung von Transposase zugänglichem Chromatin (Assay for Transposase-Accessible Chromatin using sequencing, ATAC-seq). Durch die Sequenzierung von zugänglichen Regionen im Gesamtgenom können Aussagen zur Genaktivität gemacht werden.
- Untersuchung von Histon Modifikationen auf Acetylierung und Methylierung mittels Chromatin-Immunopräzipitations-Sequenzierung (ChIP-seq). Durch eine Kombination aus einer Chromatin-Immunpräzipitation und einer Hochdurchsatzsequenzierung können Protein-DNA Interaktionsstellen kartiert werden, die im Zusammenhang mit Genexpression stehen.
Virologie/Serologie (teilweise)
Ergänzende Untersuchungen zur Virologie und Immunität werden in der Arbeitsgruppe von Prof. Dr. Sandra Ciesek in Frankfurt durchgeführt.
- Virus-Sequenzierung aus respiratorischen Proben für PatientInnen, wo dies nicht bereits im Rahmen der Diagnostik durchgeführt wurde.
- Bestimmung von SARS-CoV-2 spezifischen Antikörpern im Serum (gegen Spike und Nukleokapsid). Hier werden auch serielle Bestimmungen durchgeführt, um Aussagen zur Dauer der aktiven Immunantwort treffen zu können.*
- Bestimmung der Neutralisationsfähigkeit neutralisierender SARS-CoV-2 spezifischer Antikörper gegen die Delta-Variante.
- Bestimmung des Auftretens einer Virämie während der akuten SARS-CoV-2 Infektion. Diese Frage ist insbesondere wichtig für die Bewertung bei der Blut- und Organspende. Mittels einer hochsensitiven quantitativen PCR wird bestimmt, ob eine Virämie vorliegt und falls ja, ob und wie lange diese persistiert.*